619 951 258
Envíos gratis desde 45€
Ven a vernos
Consultas WhatsApp
Pedidos de proximidad gratuitos
Click and Collect
Envíos de proximidad gratuítos
Contacta con nosotros
Guarda productos en tu lista de deseos para comprar más tarde o compartir con tus amigos




































































































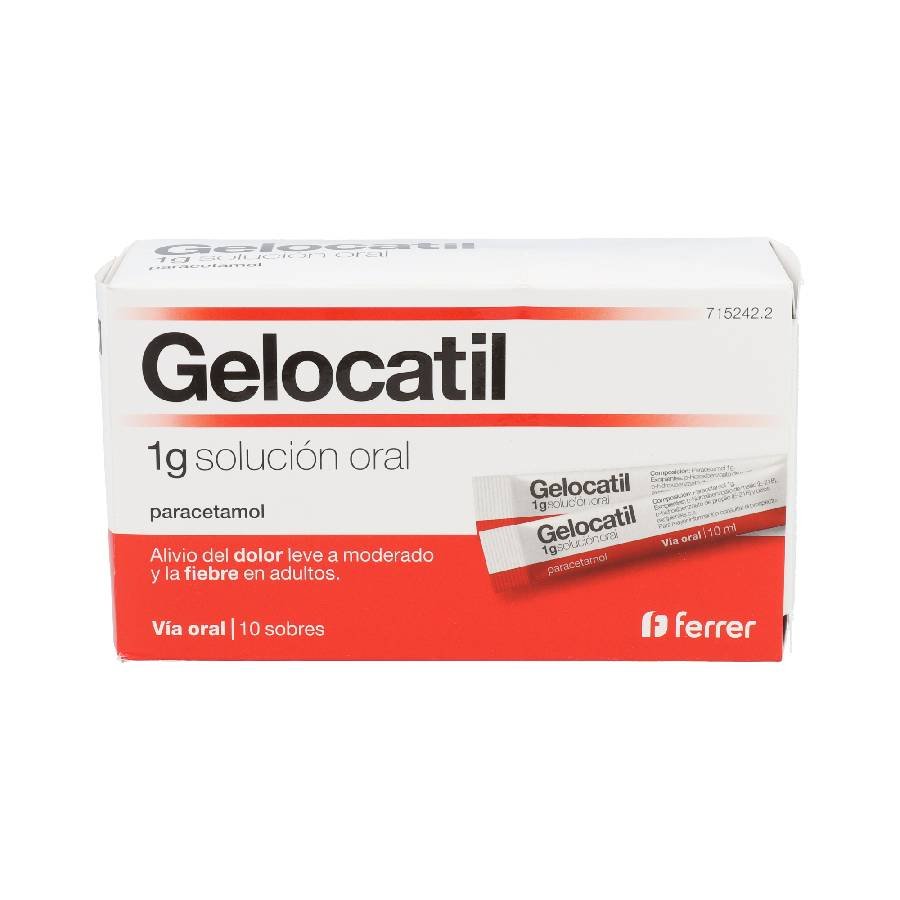
{kind=link}